
User:Ftenjo/sandbox

Uridine monophosphate synthetase (UMPS) (orotate phosphoribosyl transferase and orotidine-5'-decarboxylase) is the enzyme (EC 4.1.1.23) that catalyses the formation of uridine monophosphate (UMP), an energy-carrying molecule in many important biosynthetic pathways.[1] In humans, the gene that codes for this enzyme is located on the long arm of chromosome 3 (3q13).[2]
Structure and Function
[edit]This bifunctional enzyme has two main domains, an orotate phosphoribosyltransferase (OPRTase, EC 2.4.2.10) subunit and an orotidine-5’-phosphate decarboxylase (ODCase, EC 4.1.1.23) subunit.[3] These two sites catalyze the last two steps of the de novo uridine monophosphate (UMP) biosynthetic pathway. After addition of ribose-P to orotate by OPRTase to form orotidine-5’-monophosphate (OMP), OMP is decarboxylated to form uridine monophosphate by ODCase. In microorganisms, these two domains are separate proteins, but in multicellular eukaryotes, the two catalytic sites are expressed on a single protein, uridine monophosphate synthetase.[4]
UMPS exists in various forms, depending on external conditions. In vitro, monomeric UMPS, with a sedimentation coefficient S20,w of 3.6 will become a dimer, S20,w = 5.1 after addition of anions such as phosphate. In the presence of OMP, the product of the OPRTase, the dimer changes to a faster sedimenting form S20,w 5.6.[5][6] These separate conformational forms display different enzymatic activities, with the UMP synthase monomer displaying low decarboxylase activity, and only the 5.6 S dimer exhibiting full decarboxylase activity.[7]
It is believed that the two separate catalytic sites fused into a single protein to stabilize its monomeric form. The covalent union in UMPS stabilizes the domains containing the respective catalytic centers, improving its activity in multicellular organisms where concentrations tend to be 1/10th of the separate counterparts in prokaryotes. Other microorganisms with separated enzymes must retain higher concentrations to keep their enzymes in its more active dimeric form.[8]
Fusion
[edit]Fusion events between OPRTase and ODCase, which have led to the formation of the bifunctional enzyme UMPS, have occurred distinctly in different branches of the tree of life. For one thing, even though OPRTase is found at the N-terminus and ODCase at the C-terminus in most eukaryotes (e. g. Metazoa, Amoebozoa, Plantae and Heterolobosea), the inverted fusion, which is to say OPRTase at the C-terminus and ODCase at the N-terminus, has also been shown to exist (e. g. parasitic protists, trypanosomastids and stramenopiles). Moreover, other eukaryotic groups, such as Fungi, conserve both enzymes as separate proteins.[9]
However important the fusion order is, the evolutionary origin of each catalytic domain in UMPS is also a matter of study. Both OPRTase and ODCase have passed through lateral gene transfer resulting in eukaryotes having enzymes from bacterial and eukaryotic origin. For instance, Metazoa, Amoebozoa, Plantae and Heterolobosea have eukaryotic ODCase and OPRTase while Alveolata and stramenopiles have bacterial ones. Other rearrangements are also possible since Fungi have bacterial OPRTase and eukaryotic ODCase while kinetoplastids have the inverse combination.[9]
Merging both the fusion order and evolutionary origin, organisms end up having fused UMPS where one of its catalytic domains comes from bacteria and the other from eukaryotes.[9]
The driving force for these fusion events seems to be the acquired thermal stability. Homo sapiens OPRTase and ODCase activities lower to a greater extent when heated than the fused protein does.[10]
To determine the driving force of protein association, several experiments have been performed separating both domains and changing the linker peptide which keeps them together. In Plasmodium falciparum, the OPRTase-OMPDCase complex increases the kinetic and thermal stability when compared to monofunctional enzymes.[11] In H. sapiens, eventhough separate and fused domains have a similar activity, the former have a higher sensitivity to conditions promoting monomer dissociation.[12] Also, the linker peptide can be removed without inactivating catalysis.[13] In Leishmania donovani, separate OPRTase does not have detectable activity possibly due to lower thermal stability or lack of its linker peptide.[14]
Regulation
[edit]
UMPS is subject to complex regulation by OMP, the product of its OPRTase and the substrate for the ODCase.[15] OMP is an allosteric activator of OMP decarboxylase activity.[6] At low enzyme concentration and low OMP concentrations, OMP decarboxylase shows negative cooperativity, while at higher OMP concentrations, the enzyme shows positive cooperativity. However, when enzyme concentrations are higher, these complex kinetics do not manifest.[15] Orotate PRTase activity is activated by low concentrations of OMP,[16] phosphate,[4] and ADP.[17]
Mechanism
[edit]OPRTase
P. falciparum OPRTase follows a random pathway in OMP synthesis and degradation. Transition state analyses have used isotopic effects and quantum calculations to reveal a completely dissociated dianionic orotate structure, a ribocation and a nucleophilic pyrophosphate molecule. Nonetheless, this is unexpected since most N-ribosyltransferases involve protonated and neutral leaving groups while deprotonated orotate is not a good one in the cationic transition state.[18]
OPRTase, as a member of type I PRTases, has a prominent loop next to its active site. It is flexible in its open state and can hardly be seen in electronic density maps for some OPRTases. For catalysis to occur, a dimer must exist in which a loop from one subunit covers the active site from the other one. In Salmonella typhimurium, a new pair of antiparallel β-sheets is created and five new interatomic contacts are formed in the loop, between the loop and the rest of the protein and between the loop and the ligands.[19]
There are two possibilities as to the loop movement is concerned. It could move in a rigid manner or it could come from a disordered structure which acquires order. The second scenario seems more likely to occur in OPRTase. There must be an energy balance between the peptide new order and hydrogen bond formation in the loop, between the loop and the rest of the protein and between the loop and the ligands. There is a 30:1 equilibrium between the close and open structures in the enzyme-Mg-PRPP complex which suggest the close conformation is favored.[19]
Various roles have been proposed to the catalytic loop residues. First of all, there seems to be a correlation between the loop movement and the substrate catalysis positioning. In the biological reaction, a proton transference to the pyrophosphate (PPi) molecule could minimize negative charge accumulation eventhough the pKa for PPi is 9. Lys26, His105 and Lys103 are candidates for this transference to the α phosphate position. However, it might not be the case since lateral chains and the metal ion could neutralize some of the negative charge from the produced PPi. Transition state geometric stabilization could also be gained through loop participation.[19]
ODCase
Callahan & Miller (2007) summarize ODCase mechanisms in three proposals. The first one is the substrate carboxyl activation through electrostatic stress. The phophoryl group binding entails juxtaposition between the carboxylate group and a negatively charged Asp residue (namely Asp91 in Saccharomyces cerevisae). Repulsion between the negative charges would raise the energy value near the transition state. Nonetheless, crystallographic analyses and the lack of S. cerevisae enzyme affinity to substrate analogues where the carboxylate groups is replaced by a cationic sustituent have shown some evidence against this theory.[20]
OMP protonation on O4 or O2 before decarboxylation which entails and ilyde formation on N1 has also been considered. Proton donor absence near O4 or O2 in crystallographic structures is evidence against it along with the ilyde generation exclusion as a limiting step in 15N experiments. Moreover, doubts have aroused as to protonated intermidiate viability due to electronic stabilizers absence. As a consequence, bond rupture between C6 and C7 due to protonation of the former going through a carbanion state has been proposed.[20]
Finally, catalysis might take place by simple electrostatic attraction. C6 carbanion formation would create dipole interactions with a cationic Lys from the active site. This does not explain the velocity increase when compared with the uncatalyzed process.[20]
Clinical significance
[edit]A UMP synthetase deficiency can result in a metabolic disorder called orotic aciduria.[21]
Deficiency of this enzyme is an inherited autosomal recessive trait in Holstein cattle, and it will cause death before birth.[22]
Deficiency of the enzyme can be studied in the model organism Caenorhabditis elegans. The rad-6 strain has a premature stop codon eliminating the orotidine 5’-decarboxylase domain of the protein; this domain does not occur in any other proteins encoded by the genome. The strain has a pleiotropic phenotype including reduced viability and fertility, slow growth and radiation sensitivity.[23]
Pharmacological Importance
UMPS and its two separate domains, ODCase and OPRTase, have been shown to be essential to viability in parasites from the Chromoalveolata taxon such as L. donovani or P. falciparum.[14][24] Since UMPS, ODCase and OPRTase are different between organisms, research on species-specific inhibitors has been performed.[24][18]
Inhibition
[edit]OPRTase
Studies on OPRTase inhibition are based on substrate analogues. In Mycobacterium tuberculosis, two of the most promising inhibitors are 2,6-dihydroxipyridine-4-carboxylic acid and 3-benzylidene-2,6-dioxo-1,2,3,6-tetrahydropyridine-4-carboxylic acid. Union enthalpy and enthropy from the latter correspond to high affinity ligands. Properties such as lipophilicity, solubility, permeability and equilibrium constants are under study.[25]
Selenilation products have also been used. Abdo et al. (2010) performed reactions on 2-ethoxiethanselenic acid using electron rich aromatic substrates to produce (2-ethoxiethyl)seleno ethers. These are able to become aryl-selenilated products such as the 5-uridinyl family which has shown inhibition at submicromolar concentrations in P. falciparum and H. sapiens.[26]
ODCase
ODCase inhibitors also come from substrate analogues such as modifications on the OMP or UMP rings. In H. sapiens, ODCase has been inhibited by halide compounds derived from UMP (e. g. 5-FUMP, 5-BrUMP, 5-IUMP and 6-IUMP.[27]
In Methanobacterium thermoautotrophicum, a different strategy has been applied modifying weak interacting ligands as cytidine-5’-monophosphate which derivates into barbiturate ribonucleoside-5’-monophosphate, xantosine-5’-monophosphate.[28] P. falciparum ODCase has been succesfully inhibited by modifications on cytidine-5’-monophosphate N3 and N4.[29]
Interactive pathway map
[edit]Click on genes, proteins and metabolites below to link to respective articles.[§ 1]
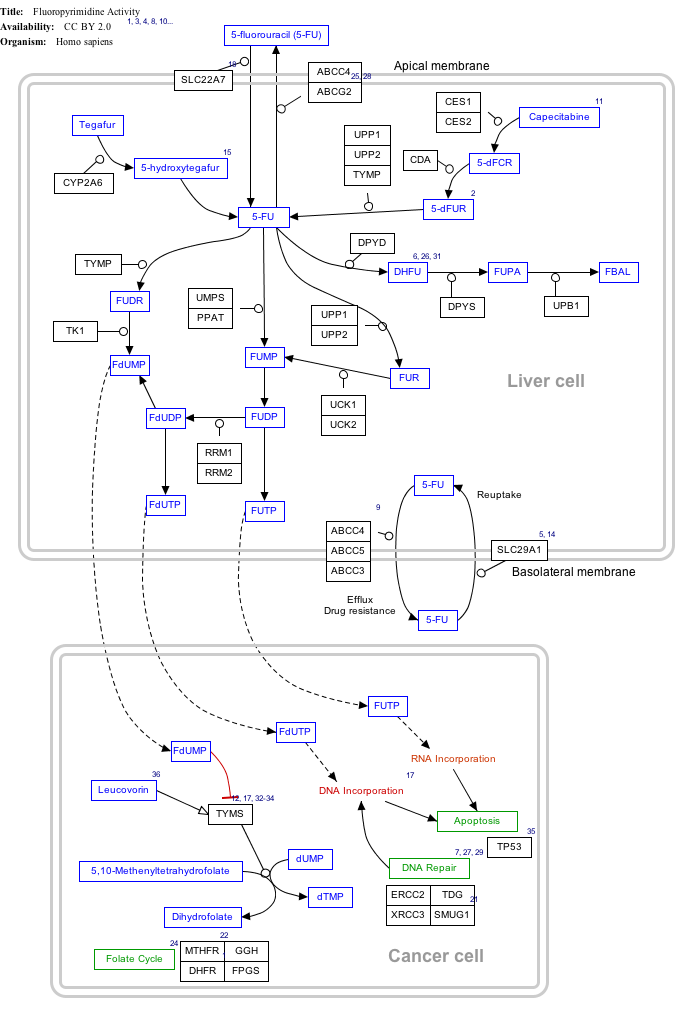
- ^ The interactive pathway map can be edited at WikiPathways: "FluoropyrimidineActivity_WP1601".
See also
[edit]References
[edit]- ^ "Entrez Gene: UMPS uridine monophosphate synthetase (orotate phosphoribosyl transferase and orotidine-5'-decarboxylase)".
- ^ Qumsiyeh MB, Valentine MB, Suttle DP (July 1989). "Localization of the gene for uridine monophosphate synthase to human chromosome region 3q13 by in situ hybridization". Genomics. 5 (1): 160–2. doi:10.1016/0888-7543(89)90103-1. PMID 2767686.
{{cite journal}}: CS1 maint: date and year (link) CS1 maint: multiple names: authors list (link) - ^ Traut, T. W.; Jones, M. E. (1996). "Uracil metabolism: UMP synthesis from orotic acid or uridine and conversion of uracil to beta-alanine: enzymes and cDNAs". Progress in Nucleic Acid Research and Molecular Biology. 53: 1–78. doi:10.1016/s0079-6603(08)60142-7. ISBN 9780125400534. PMID 8650301.
- ^ a b Jones, M. E. (1980) Annu. Rev. Biochem. 49, 253-279
- ^ Traut, T. W., and Jones, M. E. (1979) J. Biol. Chem. 254, 1143-1150
- ^ a b Traut, T. W., Payne, R. C., and Jones, M. E. (1980) Biochemistry 19, 6062-6068
- ^ Traut, T. W., and Payne, R. C. (1980) Biochemistry 19, 6068-6074
- ^ Yablonski, M. J., Pasek, D. A., Han, B., Jones. M., Traut, T. W. (1996) J. of Am. Biol. Chem. 10704-10708
- ^ a b c Makiuchi, T., Nara, T., Annoura, T., Hashimoto, T., Aoki, T. (2007). Gene. 394: 78–86
- ^ Lin, T. & Parker Suttle, D. (1995). Somatic Cell Mol. Genet. 21 (3). 161-175
- ^ Kanchanaphum, P. & Krungkrai, J. (2009). Biochem. Biophys. Res. Commun. 390. 337-341
- ^ Yablonski, M. J., Pasek, D. A., Han§, B. D., Jones, M. E., Traut, T. M. (1996). J. Biol. Chem. 271 (18). 10704–10708
- ^ Lin, T., Parker Suttle, D. (1995) Somatic Cell Mol. Genet. 21 (3). 161-175
- ^ a b French, JB., Yates, PA., Soysa, DR., Boitz, JM., Carter, NS., Chang, B., Ullman, B., Ealick, SE. (2011). J. Biol. Chem., 286 (23): 20930–20941
- ^ a b Traut, T. W. (1989) Archives of Biochemistry and Biophysics, 268, 108-115
- ^ Traut, T. W., and Jones, M. E. (1977) J. Biol. Chem. 252, 8374-8381
- ^ Chen, J.-J., and Jones, M. E. (1979) J. Biol. Chem. 254, 2697-2704
- ^ a b Zhang, Y., Deng, H., Schramm, V.L. (2010). J. Am. Chem. Soc., 132: 17023-17031
- ^ a b c Wang, G. P., Hansen, M. R., Grubmeyer, C. (2012). Biochemistry, 51: 4406−4415
- ^ a b c Callahan, B. P., Miller, B. G. (2007). Bioorg. Chem. 35. 465–469
- ^ Suchi M, Mizuno H, Kawai Y, Tsuboi T, Sumi S, Okajima K, Hodgson ME, Ogawa H, Wada Y (March 1997). "Molecular cloning of the human UMP synthase gene and characterization of point mutations in two hereditary orotic aciduria families". Am. J. Hum. Genet. 60 (3): 525–39. PMC 1712531. PMID 9042911.
{{cite journal}}: CS1 maint: date and year (link) CS1 maint: multiple names: authors list (link) - ^ Shanks RD, Robinson JL (November 1989). "Embryonic mortality attributed to inherited deficiency of uridine monophosphate synthase". J. Dairy Sci. 72 (11): 3035–9. doi:10.3168/jds.S0022-0302(89)79456-X. PMID 2625493.
{{cite journal}}: CS1 maint: date and year (link) - ^ Merry, A. (2007). Characterisation and Identification of a Radiation Sensitive Mutant in Caenorhabditis elegans. Biochemistry, Bristol. PhD.
- ^ a b Krungkrai, S. R., Aoki, S., Palacpac, N. M., Sato, D., Mitamura, T., Krungkrai, J. & Horii, T. (2004). Mol. Biochem. Parasitol., 134: 245–255
- ^ Breda, A., Machado, P., Astolfi Rosado, L., Arigony Souto, A., Santiago Santos, D., Basso, L. A. (2012). Eur. J. Med. Chem. 54: 113-122
- ^ Abdo, M., Zhang, Y., Schramm, VL., Knapp, S. (2010). Org. Lett. 12 (13). 2982-2985
- ^ Wittmann, J. G., Heinrich, D., Gasow, K., Frey, A., Diederichsen, U., Rudolph, M.G. (2008). Structure. 16. 82–92
- ^ Wu, N., Pai, E. F. (2002). J. Biol. Chem. 277 (31). 28080-28087
- ^ Purohit, M. K., Poduch, E. Wei, L. W., Crandall, I. E., To, T., Kain, K. C., Pai, E. F., Kotra, L. P. (2012). J. Med. Chem. 55 (22). 9988-9997
Further reading
[edit]PDB gallery | |
|---|---|
|



{kind=link}